


推荐产品


公司新闻/正文
「青莲客户文献」肿瘤免疫逃逸机制研究-结直肠癌TRIB3通过抑制STAT1-CXCL10轴减少CD8+ T细胞浸润并诱导免疫逃逸
人阅读 发布时间:2022-01-14 10:11
研究背景
结直肠癌中CD8+ T细胞浸润程度高提示预后良好,免疫治疗效果满意,然而,绝大多数CRC患者由于T细胞浸润不良而不能从免疫治疗中获益,T细胞转移到肿瘤组织的放松管制是肿瘤T细胞浸润缺乏的原因之一。TRIB3是一种癌蛋白,多项研究表明TRIB3在免疫应答中具有调节功能,但它在调节抗肿瘤免疫反应中的作用尚未明确。识别这些在结直肠癌中的免疫改变及其潜在机制,可能为改善可能受益于ICB治疗的结直肠癌患者的频率提供新的可能性。研究过程
1、探究TRIB3是否参与肿瘤免疫应答的调节
2、证明糖尿病相关的TRIB3表达与CRC组织中CD8+ T细胞的排除有关
3、确定TRIB3通过STAT1-CXCL10信号通路抑制T细胞浸润
4、探究糖尿病中的高糖状态是如何诱导TRIB3表达
5、研究互作蛋白乙酰化如何作为TRIB3稳定信号(相互作用蛋白-质谱鉴定-青莲百奥提供技术支持)
6、确证TRIB3-K240乙酰化介导CRC中高糖诱导的免疫逃逸
1、探究TRIB3是否参与肿瘤免疫应答的调节
首先构建MC38细胞皮下接种免疫缺陷BALB/c-nude小鼠或免疫正常C57BL/6J小鼠并敲除TRIB3,发现敲除TRIB3可降低MC38肿瘤的生长。MC38细胞中TRIB3的缺失导致约50%的肿瘤抑制率,表明TRIB3在抗肿瘤免疫应答调控中的潜在作用。MC38细胞中TRIB3的敲除增加了肿瘤CD8+ T细胞浸润,过度的TRIB3 MC38细胞促进肿瘤的生长和抑制CD8 + T细胞浸润和功能。研究结直肠癌小鼠模型的组织切片,TRIB3的下调增强了结直肠癌组织中CD8+ T细胞的浸润,相比之下,TRIB3的异位表达抑制了浸润。同时CRC患者的肿瘤组织芯片证实TRIB3蛋白表达与CD8+ T细胞浸润呈负相关。总而言之,TRIB3的高表达会损害CRC组织中CD8+ T细胞的浸润。

2、证明糖尿病相关的TRIB3表达与CRC组织中CD8+ T细胞的排除有关
糖尿病与早发性CRC风险增加相关,因此作者发现糖尿病CRC肿瘤组织中TRIB3在高度表达,糖尿病和CRC患者的肿瘤组织中CD8+ T细胞浸润与非糖尿病患者相比减少,且TRIB3的表达与CD8+ T细胞浸润CRC组织呈负相关。与糖尿病小鼠相比,非糖尿病小鼠中有更多的CD8+ T细胞浸润MC38细胞来源的肿瘤组织。将对照组或TRIB3缺陷的MC38细胞接种到糖尿病小鼠,评估TRIB3与糖尿病肿瘤组织中CD8+ T细胞的排除有关。对肠道上皮特异性TRIB3缺陷老鼠(TRIB3IEC-KO)建立CRC糖尿病条件下小鼠模型,发现细胞特异性TRIB3的缺失减缓了自发CRC进展。这些数据表明,糖尿病相关的TRIB3表达阻碍了CD8+ T细胞向CRC组织的浸润。
3、确定TRIB3通过STAT1-CXCL10信号通路抑制T细胞浸润
趋化因子是影响CD8+ T细胞转移到肿瘤部位的主要因素,探究了了TRIB3是否通过影响趋化因子的表达来减少T细胞的肿瘤浸润。研究表明敲除TRIB3可上调CXCL10的表达,ELISA实验证实在正常或高糖处理条件下,敲除TRIB3会增加CXCL10蛋白的表达,高糖条件下CXCL10蛋白的减少由于TRIB3缺失而被逆转,过表达TRIB3抑制了CXCL10蛋白的表达,阻断CXCL10逆转了TRIB3诱导的CD8+ T细胞在人原代CRC-30和小鼠MC38细胞中的迁移。
STAT1为介导CXCL10转录的转录因子,参与了TRIB3调控的CXCL10转录。双荧光素酶报告基因检测显示,在MC38细胞中,TRIB3过表达会下调STAT1活性,TRIB3缺失促进了STAT1的转录,但没有改变其蛋白的稳定性。紧接着建立了CXCL10过表达的MC38衍生肿瘤模型确定TRIB3是否通过调节CXCL10在体内损害CD8+ T细胞浸润。CXCL10的异位表达抑制了TRIB3过表达的原生效应,消除了TRIB3对CD8+ T细胞浸润的抑制作用,表明TRIB3在一定程度上通过抑制CXCL10介导的CD8+ T细胞招募来促进肿瘤进展。使用糖尿病CXCL10基因敲除(CXCL10KD)的mc38衍生肿瘤模型,进一步验证了这一结论。

4、P300诱导TRIB3乙酰化,增强了TRIB3蛋白的稳定性
糖尿病中的高糖状态是如何诱导TRIB3表达的?研究发现高糖处理对TRIB3的转录没有影响,但增强了外源转染的细胞中TRIB3的蛋白表达。在HCT-8细胞中高糖处理也延长了TRIB3的半衰期,表明高糖通过增强TRIB3蛋白的稳定性促进了TRIB3的高表达。
高葡萄糖导致乙酰辅酶CoA的产生,也是蛋白质乙酰化修饰所需的乙酰基的供体。因此,研究了高糖是否诱导了TRIB3的乙酰化改变。高糖处理以时间依赖的方式刺激HCT-8、DLD-1和HT-29细胞中的TRIB3乙酰化,利用siRNA介导的功能缺失策略筛选了经典的乙酰转移酶TIP60、P300、CBP。后续发现P300敲除或C646处理降低了4T1细胞中TRIB3的表达,在CRC患者的组织中,肿瘤组织样本中的P300高于相邻的非肿瘤组织样本。这些结果表明P300诱导了CRC中TRIB3的乙酰化,并增强了TRIB3蛋白的稳定性。GST免疫共沉淀实验证实了TRIB3和P300之间的直接相互作用。随后作者将TRIB3 KDC结构域的所有赖氨酸残基突变为精氨酸(R)残基,确定K240是P300对TRIB3的乙酰化位点,表明p300介导的K240乙酰化作用是维持TRIB3稳定性的信号。
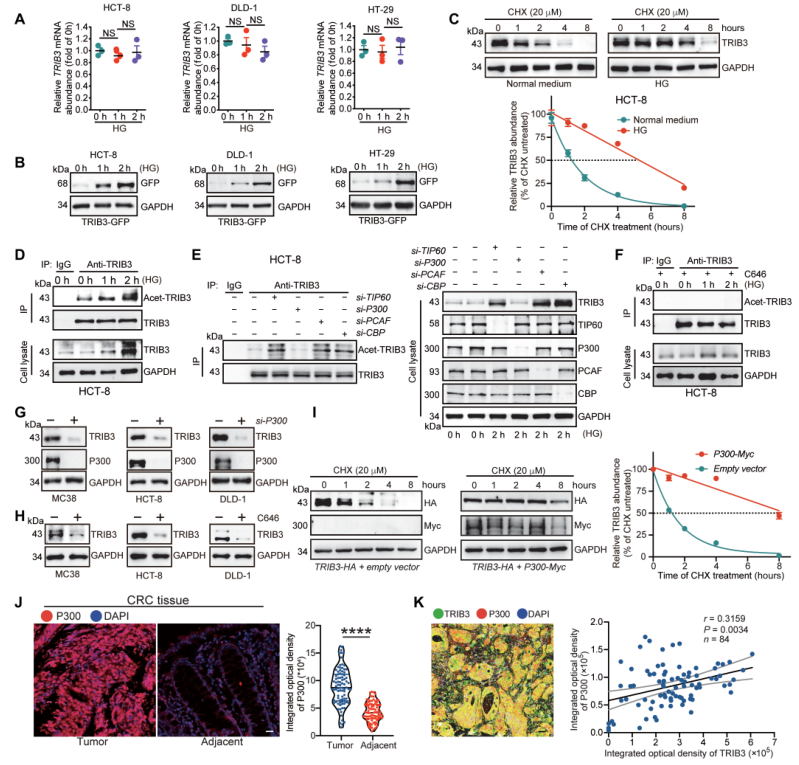
5、TRIB3的乙酰化削弱了其与SIAH1 E3连接酶的相互作用接下来研究了p300介导的K240乙酰化如何作为TRIB3稳定信号,过表达P300抑制了野生型突变体泛素化,但对TRIB3-K240R没有抑制。通过免疫沉淀和质谱分析,发现CHIP蛋白TRIM38和C末端被鉴定为TRIB3的结合部位。P300的影响与四个TRIB3 E3连接之间的相互作用,尤其P300的交互抑制TRIB3 SIAH1。此外,当SIAH1基因缺失时,P300并没有增强TRIB3的稳定性。总之,这些数据表明p300催化的TRIB3的K240乙酰化阻止了SIAH1结合和TRIB3泛素化降解。

6、TRIB3-K240乙酰化介导CRC中高糖诱导的免疫逃逸
为了确定TRIB3 K240乙酰化在CRC免疫应答中的关键作用,研究人员构建了K240乙酰化抗性MC38和HCT-8细胞(TRIB3KD/K240ROE,以下简称TRIB3Ace-Resis)。TRIB3Ace-Resis细胞表现出更高的CXCL10表达。TRIB3Ace-Resis组TRIB3降低,增加pSTAT1和STAT1表达,CXCL10的转录趋势与STAT1相同。这些数据表明,P300在K240位点对TRIB3进行乙酰化修饰,通过TRIB3-STAT1 - CXCL10轴,在非糖尿病和糖尿病性结直肠癌情况下,都是一种关键的分子机制。

7.靶向TRIB3乙酰化使肿瘤对ICB治疗敏感
为了探索靶向TRIB3乙酰化在体内肿瘤免疫和免疫治疗中的潜力,使用C646在患有CT26原位CRC肿瘤的糖尿病小鼠中药理抑制TRIB3乙酰化。单药治疗发现部分减缓肿瘤进展,联合抗体抗肿瘤效果增强,增加了CD8+ T细胞浸润,并增加了肿瘤微环境中IFN-+和颗粒酶B+ CD8+ T细胞的百分比。这些数据表明,通过药物抑制TRIB3乙酰化,结合ICB疗法,通过降低TRIB3稳定性,招募和激活肿瘤微环境中的CD8+ T细胞,从而诱导有效的抗肿瘤免疫。

小结
此项研究证明了TRIB3在各种CRC小鼠模型中抑制CD8+ T细胞浸润。发现TRIB3被乙酰转移酶P300乙酰化,抑制了TRIB3的泛素化和随后的蛋白酶体降解。异位表达的TRIB3通过增强表皮生长因子受体信号通路,抑制信号转导和转录激活因子1 (STAT1)的激活和STAT1介导的CXCL10转录,导致肿瘤浸润T细胞的减少。这些发现证实了TRIB3是CRCs中CD8+ T细胞浸润的负调节因子,突出了治疗免疫“冷”CRCs的潜在治疗靶点。
询价列表

暂时没有已询价产品
