


推荐产品


公司新闻/正文
「青莲聚焦」Cell Metabolism综述:铁死亡的代谢基础
人阅读 发布时间:2022-04-22 11:22

生物体生长发育及众多生理病理过程中,细胞程序性死亡必不可少。它属于细胞可调节性死亡的范畴,过度会对组织细胞造成不可逆性损伤,适度则可清除受损细胞起到修复和维持自身稳态的作用。除经典的细胞凋亡,越来越多的细胞程序性死亡形式逐一被发现,例如坏死性凋亡、自噬、细胞焦亡、铁死亡等,其中铁死亡在形态学、遗传学和生物化学上不同于其他形式,主要表现为氧化还原体系失衡,脂质过氧化物的过度蓄积等。是一种在铁离子的参与下,因铁依赖性活性氧(ROS)的异常堆积而导致氧化还原平衡失调,导致细胞死亡的新型死亡方式。接下来这篇综述,描述了细胞内几种可直接影响细胞对脂质过氧化和铁死亡敏感性的代谢途径,如硫醇代谢、脂肪酸代谢、甲羟戊酸途径和线粒体呼吸等。并总结了目前发现的主要的铁死亡抑制系统。之后,作者还讨论了铁死亡在人类疾病发展预防治疗方面的应用。

标题:The Metabolic Underpinnings of Ferroptosis.
期刊:Cell Metabolism (IF =21.567)
时间:2020.12
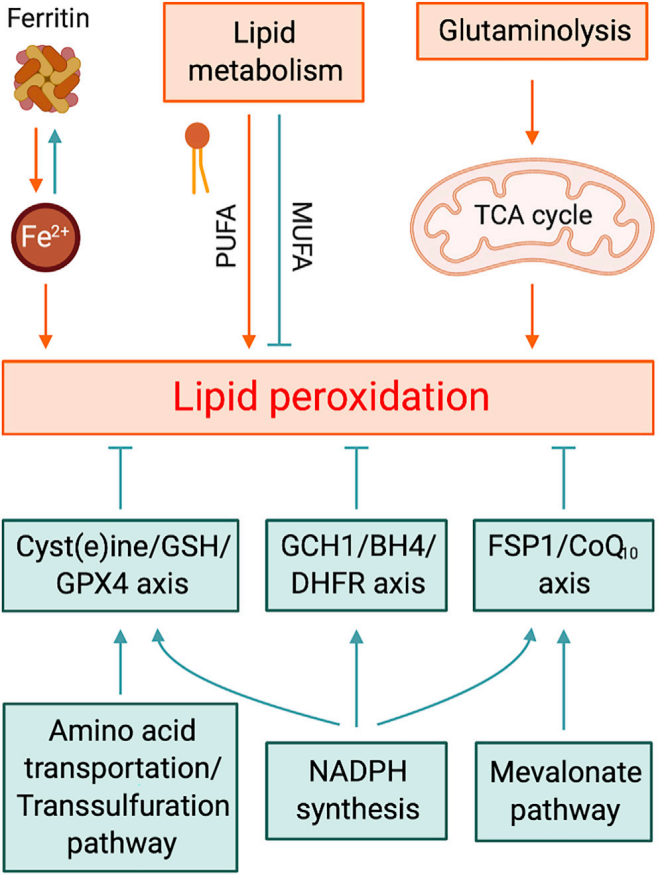
影响细胞对铁死亡易感性的代谢过程
一
硫醇依赖性氧化还原系统
含巯基的GSH是哺乳动物细胞中的主要抗氧化剂,也是GPX4的底物,因此GSH供应不足将直接影响GPX4发挥功能,从而使细胞发生铁死亡。半胱氨酸(cysteine)是合成GSH的原料,主要由胱氨酸(cystine)还原而来,而胱氨酸进入细胞则依赖胱氨酸/谷氨酸逆转运蛋白SLC7A11(也写作system xc-)。因此,SLC7A11被视作铁死亡的关键的上游调节因子之一。近年来研究发现,包括p53在内的一些肿瘤抑制基因以及干扰素γ被发现能够抑制SLC7A11相关的表达和转运活性。另一方面,肿瘤细胞也可利用与压力相关的转录因子(如ETS-1、ATF4、NRF2等)上调SLC7A11的表达来抵抗铁死亡。食物中的蛋氨酸也可以通过转硫途径在一定程度上为细胞供应半胱氨酸。在一些肿瘤细胞中,转硫途径甚至可以不断被激活。经erastin(SLC7A11抑制剂)给药数天的卵巢癌细胞可逐渐增强转硫作用以弥补胱氨酸的缺乏。新的研究证实,破坏转硫途径相关酶均可延缓肿瘤生长。
二
细胞内铁代谢
铁死亡自发现以来,围绕铁代谢紊乱和脂质过氧化这两大基本特征展开了大量研究,因此顾名思义,铁死亡中的脂质过氧化和细胞膜破裂需要铁的参与。所以铁螯合剂可以抑制铁死亡,而通过纳米颗粒将铁装入肿瘤细胞中可诱导肿瘤细胞发生铁死亡。在生理条件下,细胞铁的吸收主要由质膜蛋白转铁蛋白受体(TFR1)控制,该蛋白通过受体介导的内吞作用携带转铁蛋白结合的铁进入细胞,因此TFR1被认为是铁死亡发生的标志蛋白,通过敲除 TFR1 来阻断这一过程可防止铁死亡。细胞的铁存储在铁蛋白中,其蛋白量特别是重链的表达减少会增加铁池与细胞对铁死亡的敏感程度。相反,通过抑制铁蛋白的自噬性降解则能使细胞抵抗铁死亡。目前认为细胞中铁的输出由质膜上与多铜铁氧化酶(如铜蓝蛋白)偶联的铁输出蛋白FPN1主宰。另外,含有铁蛋白的多泡小体和外泌体也可以在一定程度上向细胞外排铁。其他与铁相关的蛋白包括多聚胞嘧啶结合蛋白PCBP1和血红素氧合酶HO-1也参与了铁死亡的调节。PCBP1负责铁的运送,有研究报道小鼠肝细胞特异性缺失PCBP1导致铁增加,并加剧肝脏中的铁死亡。但目前为止学者们并未对HO-1在铁死亡中的作用达成共识。近年来,GSH在调节细胞内铁稳态中的作用引起了广泛的关注。结合于GSH的亚铁离子被认为是铁池中铁的主要形式。铁与PCBP1的结合也依赖于GSH。
三
甲羟戊酸途径
甲羟戊酸途径生成的异戊烯焦磷酸(IPP)、 角鲨烯、CoQ10和胆固醇能够从各个方面影响铁死亡。其中IPP是角鲨烯和CoQ10的前体。阻断该途径限速酶的他汀类药物会干扰GPX4的有效翻译,从而增加细胞对铁死亡的敏感性。有研究提出角鲨烯也能被癌细胞当成抵抗铁死亡的有力工具。但角鲨烯合成酶SQS的激动剂FIN56却能够诱导铁死亡,这可能是CoQ10消耗所致,后者被证实在铁死亡进程中参与保护机制。有意思的是, 基因筛选数据显示,之前为人们所知的线粒体相关凋亡诱导因子AIFM2,通过利用CoQ10阻止脂质自氧化而跻身为继GPX4之后的第二大铁死亡抑制因子阵营,并由此被重新命名为铁死亡抑制蛋白FSP1。基于胆固醇在真核细胞质膜中的普遍存在及其对羟基自由基等氧化剂的内在敏感性,人们也被认为胆固醇参与了铁死亡。GPX4是已知的能够直接清除胆固醇过氧化物的酶,GPX4活性降低会加剧外源性胆固醇诱导的细胞死亡,而GPX4的过表达能极大地提高细胞对过氧化胆固醇的抵抗力。但实际上并没有确凿证据能够证实胆固醇的氧化真的助力铁死亡。相比之下,胆固醇的前体,7-脱氢胆固醇的氧化还原活性甚至更胜一筹,它的经典自由基氧化反应速度比胆固醇和花生四烯酸(AA)分别高出200倍和20倍。但无论胆固醇抑或是7-脱氢胆固醇,它们对脂质过氧化和铁死亡的潜在调节作用仍需进一步探索。
四
线粒体在铁死亡中的角色
线粒体在铁死亡中的作用仍是有争议的,学者们认为,线粒体可能通过其他未知因素或者代谢途径参与铁死亡。有研究指出,线粒体促进铁死亡的作用可能归因于线粒体呼吸产生的活性氧 (ROS),使得线粒体呼吸链复合酶受损,但是为什么线粒体呼吸会失控?一方面,SLC7A11抑制或胱氨酸剥夺可导致GSH锐减,从而削弱其对ROS的清除能力。另一方面,SLC7A11的失活可导致细胞内谷氨酸的蓄积,并转化生成α-酮戊二酸,后者通过促进TCA循环,导致线粒体膜电位超极化。即线粒体呼吸作用紊乱是抗氧化系统和由谷氨酰胺分解助燃的TCA循环系统这两大势力此消彼长的结果。而RSL3并不促进谷氨酰胺分解,因此线粒体对于RSL3诱导的铁死亡不起作用。
五
NADPH—预防铁死亡
NADPH的基础水平被认为是几种癌细胞系中铁死亡敏感性的生物标志物,NADPH的匮乏将促进细胞发生铁死亡。但NADPH也可被NADPH氧化酶利用产生超氧阴离子,对这一过程的激活或抑制也可能决定细胞是否发生铁死亡。同时许多途径都可生成NADPH,例如磷酸戊糖途径(PPP)、NADH磷酸化途径以及从异柠檬酸转化的α-酮戊二酸途径等。以上途径只有磷酸戊糖途径正向调控铁死亡,其余二者则反向调控。因此,尽管研究更倾向于认为NADPH的抗铁死亡作用要胜过其促铁死亡作用,但NADPH实际可能在铁死亡调控中发挥双重作用。
六
多不饱和脂肪酸(PUFA)
初研究认为含花生四烯酸(AA)或肾上腺酸的磷脂酰(PE)是铁死亡中的磷脂/脂质过氧化的主要底物,而后续的数据则指向范围更广的多种PUFA。操纵PUFA的合成或降解过程将影响细胞对铁死亡的敏感性。目前在铁死亡中脂质过氧化作用影响的主要是酯化的PUFA,而非游离的PUFA。因此,减少长链脂肪酸水解酶ACSL4(ACSL4优先催化长链脂肪酸成酰基辅酶A酯)与使AA重新酰化成溶血磷脂的LPCAT3将显著增加细胞的铁死亡抗性。过氧化物酶体是合成多不饱和醚磷脂的位点,因此也能使细胞对铁死亡敏感。与PUFA酯化相关的还包括低氧诱导的脂滴相关蛋白HILPDA、磷脂酶PLA2和溶血磷脂酰丝氨酸脂酶ABHD12等,均展示出对铁死亡正向和负向调控的作用。除了靠PLA2和ABHD12水解磷脂上的PUFA,细胞还具备另一种避免脂质过氧化的途径—利用外源性单不饱和脂肪酸(MUFA)取代磷脂PUFA。研究显示,突变的KRAS肺癌细胞和淋巴中的黑色素瘤细胞的存活和增殖均依赖于ACSL3。上述研究一致认为,磷脂中的PUFA充足是铁死亡插手细胞命运的先决条件。
七
铁死亡的标志—脂质过氧化
磷脂PUFA氧化物(PLOOH)是如何引发铁死亡一直存在争论。主要针对的是脂质过氧化究竟是酶催化的还是非酶促的自氧化?酶促的研究强调脂氧合酶LOX家族(尤其是ALOX15)在脂质过氧化和铁死亡中起核心作用。非酶促项研究质疑了LOX 在铁死亡起始过程中的主要作用。首先,敲除ALOX15并不能挽救GPX4缺失引起的铁死亡。其次,对多种LOX亚型(5-LOX、p12-LOX和15-LOX-1)过表达的研究指出,无论LOX对PLOOH的初始积累有何贡献,脂质的非酶促自氧化才是铁死亡的真正诱因。主张非酶促驱动的研究称,氢过氧化物(LOOH)的临界水平(约20 μM)可能是多种细胞系中铁死亡发生的阈值。但酶促反应对于诱导铁死亡的作用仍然是毋庸置疑的,因为如果没有酶促的贡献,PLOOH就可能无法达到临界值,尤其细胞内还具备强大的抗氧化剂体系以随时清除PLOOH。非酶促派也必须承认,LOX某些亚型或细胞色素P450氧化还原酶(POR)的缺失的确可以明显缓解GPX4缺乏引起的不良后果。铁积极参与脂质过氧化的启动,但是它如何促成这一过程仍然难以评估。含铁酶促的活性依赖铁的参与,铁也可以通过芬顿反应促进脂质的非酶促自氧化。总之不管哪种方式,铁螯合剂都是阻挡脂质过氧化和铁死亡的主要手段。
主要的铁死亡抑制机制
一
Cyst(e)ine/GSH/GPX4机制
多年来,Cyst(e)ine/GSH/GPX4系统被认为是调节铁死亡的机制。抑制胱氨酸-谷氨酸逆向转运或GSH的生成、降低GPX4表达水平或活性均可直接影响铁死亡的发生发展。GPX4作为一种硒蛋白,其生物合成依赖于硒代半胱氨酸的共翻译结合。
二
NAD(P)H/FSP1/CoQ10机制
FSP1被认为在某些特定条件下具有促凋亡作用,FSP1可经细胞质或线粒体转位进入细胞核,从而触发DNA降解。近些年的研究显示,FSP1具有平行于Cyst(e)ine-GSH-GPX4轴的抗铁死亡机制。FSP1通过NAD(P)H,还原CoQ10,进而还原脂质自由基,从而抑制脂质过氧化。
三
GCH1/ BH4/DHFR 机制
科研人员通过CRISPR全基因组筛选并确定了一种不依赖于GPX4的铁死亡抑制:GCH1/ BH4/DHFR 机制。BH4是一种自由基捕获抗氧化剂,但它的回收再利用需要二氢叶酸还原酶DHFR的参与,因此如果阻断二氢叶酸还原酶DHFR,则可协同GPX4抑制剂发挥诱导铁死亡的作用。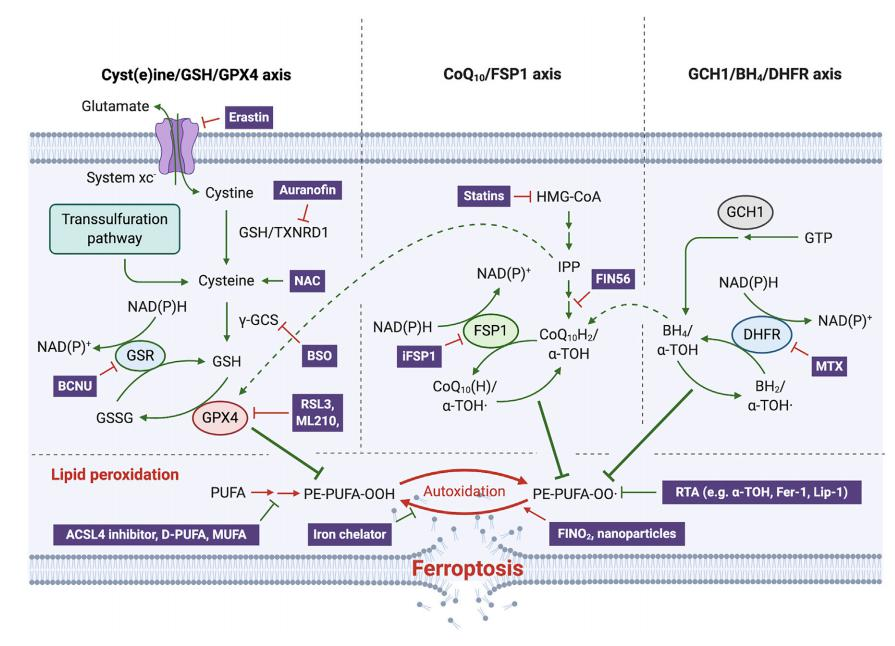
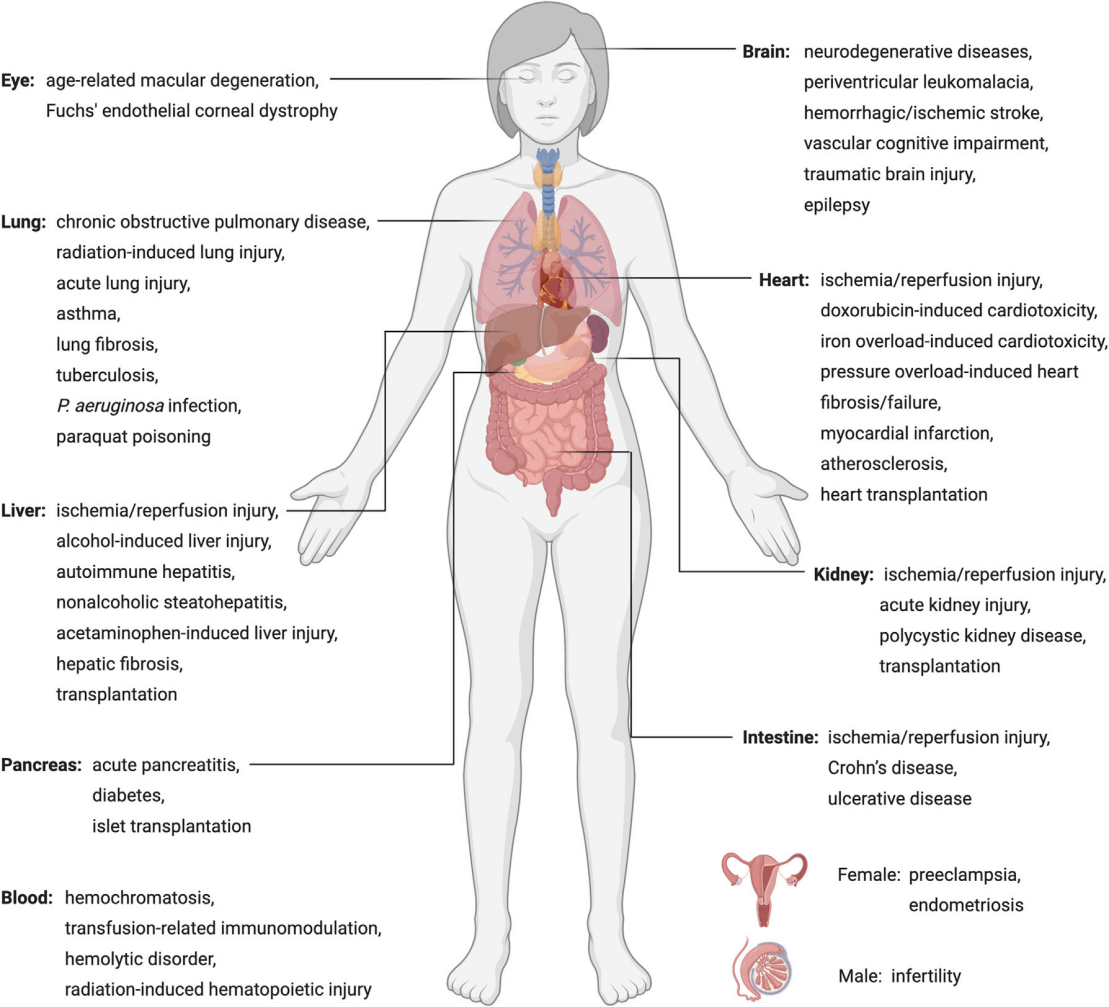
铁死亡在人类疾病中的意义
由于缺乏针对铁死亡的强有力的生物标记物,铁死亡和人类疾病之间的联系仍然没有得到充分的探索。因此,目前对这一方向的理解主要是基于使用条件gpx4敲除小鼠和再现铁死亡特征的病理模型的研究。对疾病模型中铁死亡抑制剂的治疗特征与治疗效果的验证有助于理解铁死亡在某些疾病环境中的参与。铁死亡诱导剂已被证明在杀死几种类型的癌症方面特别有效。在接受免疫治疗、放疗和化疗的癌症患者中也检测到了铁死亡现象。因此,诱导性铁死亡可以作为治疗这些难治性癌症的新方法。铁死亡诱导剂已被证明可以增强辐射的抗肿瘤作用,但如何调节铁死亡通路,从而在放射治疗中获得较大的临床效益仍是一个需要进一步研究的关键问题。
END
询价列表

暂时没有已询价产品
